Logiciel Norns : Interface utilisateur permettant de spécifier l’extraction de manière graphique.
Copyright : GREYC/CERMN
Article publié sur le site du CNRS
La mise sur le marché d’un nouveau médicament est le fruit d’un processus de recherche très long et complexe. Pour toucher la cible que l’on souhaite atteindre chez les patients, il est en effet nécessaire de trouver la « clé » qui lui correspond. Cette « clé » correspond à une organisation spatiale précise d’atomes, appelée motif structural d’une molécule. Mais chaque molécule peut contenir un grand nombre de motifs structuraux ! Des enseignants-chercheurs du Groupe de REcherche en Informatique, Image, Automatique et Instrumentation de Caen (GREYC – CNRS/ENSICAEN/Université Caen Normandie) et du Centre d’Études et de Recherche sur le Médicament de Normandie (CERMN – CNRS/INC3M/Université Caen Normandie) ont développé un logiciel permettant de trouver automatiquement des motifs structuraux favorables à un effet thérapeutique donné.
Habituellement, la conception d’un médicament commence par l’identification de cibles chez le patient (protéines-acides nucléiques classiquement) dont on souhaite moduler l’activité. Il faut déterminer, par la suite, un ensemble de molécules qui interagissent suffisamment avec ces cibles : les molécules candidates. Parmi ces dernières, la molécule qui maximise l’effet thérapeutique, tout en limitant les effets secondaires et les effets indésirables, sera la plus intéressante et sera sélectionnée pour les études précliniques. La mesure de ces propriétés se faisant expérimentalement, le processus consomme de nombreuses ressources. La réalisation de toutes ces étapes nécessite d’énormes investissements, pour un résultat très incertain. Ce coût inhérent constitue un facteur qui limite énormément les innovations pharmaceutiques.
Dans ce contexte, des chercheurs du GREYC et du CERMN ont développé un logiciel permettant de déterminer de façon automatique les motifs structuraux, c’est-à-dire l’organisation spatiale souhaitée des atomes, en fonction de l’objectif thérapeutique recherché. Ces motifs structuraux peuvent ensuite être directement utilisés comme critères dans la sélection des molécules candidates. Une sélection automatique efficace permet ainsi aux chimistes médicinaux de concentrer rapidement leurs efforts sur les molécules les plus prometteuses.
La technologie de découverte de ces motifs structuraux s’appuie sur des notions et des techniques de fouille de données (data mining), et plus spécifiquement, de fouille de graphes. En informatique, un graphe modélise une relation : dans une molécule, on considère que les atomes sont en relation grâce aux liaisons interatomiques. Pour trouver le résultat, le logiciel parcourt intensivement les données, en l’occurrence des molécules, chacune modélisée par un graphe.
Afin de faire fonctionner les algorithmes de découverte des motifs structuraux discriminants, le jeu de données est séparé en deux sous-ensembles : les molécules actives et les inactives. Ensuite, les algorithmes vont construire des hypothèses, sur les motifs structuraux, de plus en plus précises, en gardant une trace des molécules dans lesquelles les motifs structuraux apparaissent. Les plus intéressants seront gardés comme résultats s’ils apparaissent beaucoup plus souvent dans les molécules actives.
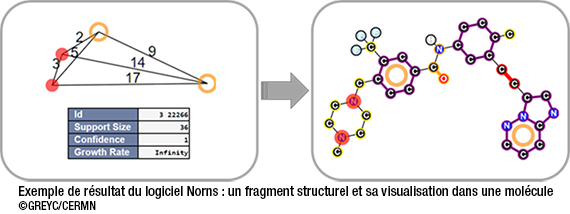
Toutefois, même en mettant des conditions exigeantes sur les différences de fréquence d’apparition de ces motifs, le calcul produit souvent un nombre de possibilités très important, dont beaucoup n’ont pas tout à fait capturé le phénotype, par exemple la relation entre la structure et l’impact biologique. Afin de parcourir aisément la grande quantité de résultats, d’évaluer les motifs structuraux et de sélectionner les plus pertinents, les chercheurs ont placé les différentes possibilités dans un diagramme de Hasse. Cette structure permet à l’expert de naviguer entre les motifs discriminants en modifiant les contraintes structurelles et d’accéder aux molécules testées qui satisfont la contrainte structurale en cours. De plus, la navigation s’accompagne de mesures de qualité, ces dernières allant de la très naturelle fréquence d’apparition à des mesures plus sophistiquées comme la stabilité [1]. La stabilité d’un motif structural quantifie sa dépendance aux petits changements dans les données : un motif est stable si son intérêt ne repose pas uniquement sur quelques mesures biologiques.
Par ailleurs, afin de trouver les possibilités expliquant au mieux les raisons d’un phénotype, les chercheurs ont également développé des algorithmes qui fonctionnent sur des représentations sophistiquées des molécules. Ils ont par exemple conçu un algorithme permettant de calculer les motifs discriminants lorsque les molécules sont représentées par plusieurs conformations spatiales. En effet, la structure d’une molécule n’étant pas figée, elle peut s’envisager comme une suite d’états plus ou moins stables, chaque état étant représenté par un graphe spatial [2].
Ce type de calcul peut s’appliquer sur d’autres données “omiques”. Les chercheurs ont par exemple exploité des données issues de relevés métabolomiques. Il s’agit de données cliniques où, pour un patient donné à un instant donné, ces mesures indiquent la concentration de centaines de métabolites dans le sang. Leur étude dans le cas du cancer du foie a permis de montrer qu’il existe des concentrations de métabolites corrélées au cancer du foie, même si ces dernières varient selon le patrimoine génétique du patient [3]. Cette technique permettra certainement à l’avenir des applications dans le cadre d’une pratique personnalisée de la médecine.
[1] Jean-Philippe Métivier, Alban Lepailleur, Aleksey Buzmakov, Guillaume Poezevara, Bruno Crémilleux, Sergei O. Kuznetsov, Jérémie Le Goff, Amedeo Napoli, Ronan Bureau, Bertrand Cuissart, Discovering Structural Alerts for Mutagenicity Using Stable Emerging Molecular Patterns. Journal of Chemical Information and Modeling 55(5) : 925-940, 2015, DOI : 10.1021/ci500611v
[2] Julien Rabatel, Thomas Fannes, Alban Lepailleur, Jérémie Le Goff, Bruno Crémilleux, Jan Ramon, Ronan Bureau, Bertrand Cuissart : Non a priori automatic discovery of 3D chemical patterns — application to mutagenicity, Molecular Informatics, DOI : 10.1002/minf.201700022, published online 7 June 2017
[3] Guillaume Poezevara, Sylvain Lozano, Bertrand Cuissart, Ronan Bureau, Pierre Bureau, Vincent Croixmarie, Philippe Vayer, Alban Lepailleur : A Computational Selection of Metabolite Biomarkers Using Emerging Pattern Mining — a Case Study in Human Hepatocellular Carcinoma, 16 (6), pp 2240–2249, 2017, DOI : 10.1021/acs.jproteome.7b00054